Інститут технологій Массачусетса (MIT) презентував новаторський додаток ChemXploreML, що значно спрощує прогнозування властивостей молекул для хіміків. Відтепер науковці можуть робити важливі прогнози без потреби в глибоких програмістських навичках, що відкриває нові можливості для досліджень у галузі медицини та матеріалознавства.
Доступність та ефективність
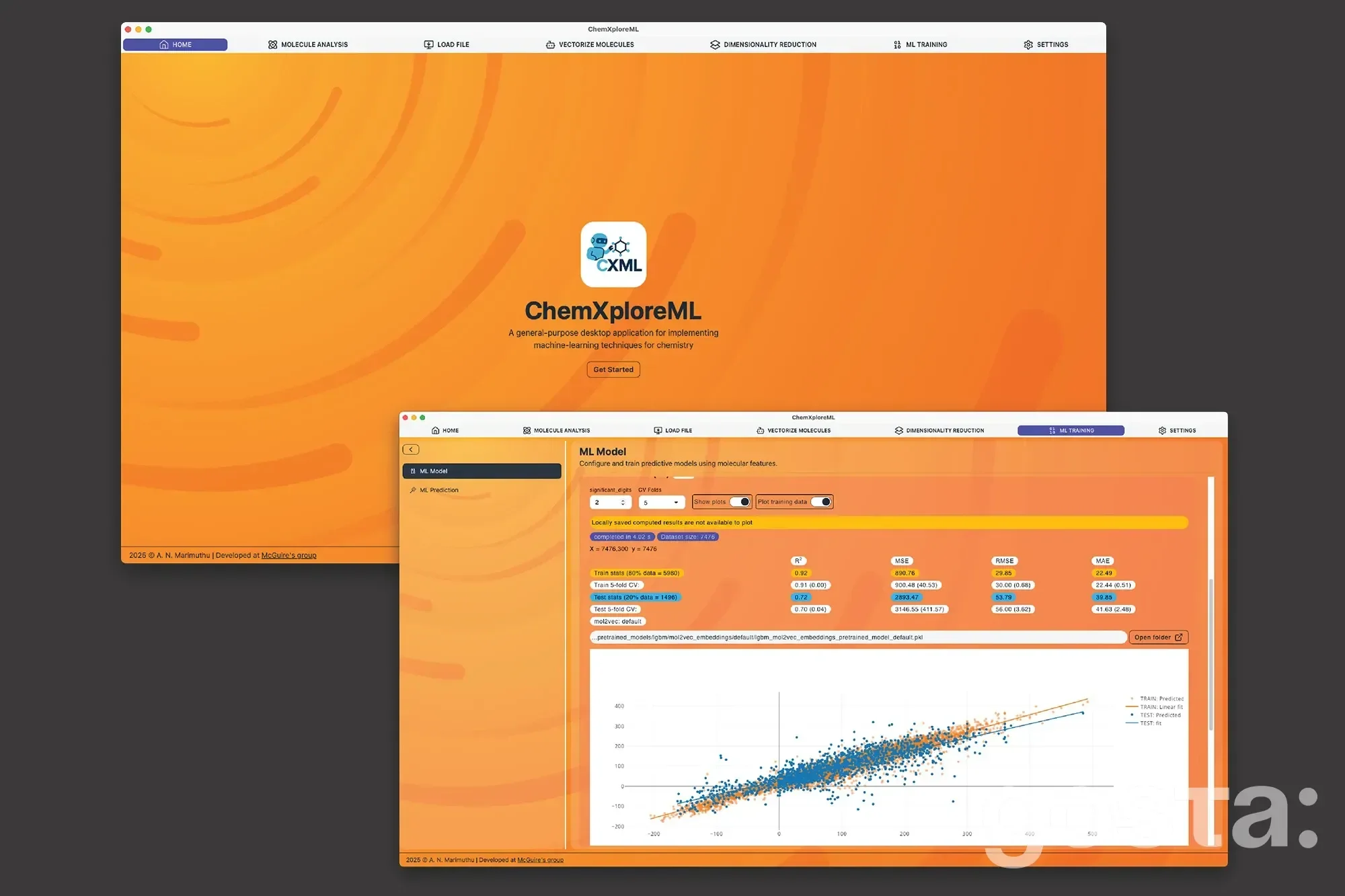
Одним з ключових викликів в області хімічного машинного навчання є перетворення молекулярних структур у цифрову мову. ChemXploreML автоматизує цей складний процес за допомогою вбудованих "молекулярних ембеддерів", які переводять хімічні структури у числові вектори. Завдяки передовим алгоритмам, програмне забезпечення точно прогнозує властивості, такі як температура кипіння та плавлення, через інтуїтивний графічний інтерфейс.
Цей додаток, доступний для завантаження на популярні платформи, працює повністю офлайн, що забезпечує конфіденційність дослідницьких даних. Він був протестований на декількох ключових молекулярних властивостях органічних сполук і показав високу точність.
Майбутнє наукових досліджень
Завдяки ChemXploreML, хіміки можуть легко налаштовувати та застосовувати машинне навчання для вирішення унікальних викликів, від розробки стійких матеріалів до вивчення складної хімії міжзоряного простору. Це відкриває нові горизонти для інновацій у хімічних науках, роблячи процес скринінгу швидшим і дешевшим.
